Olivier Hyrien
Eukaryotic Chromosome Replication
Our long-term goal is a quantitative understanding of whole-genome replication in multiple eukaryotes.

Research context
DNA replication is tightly regulated to accurately duplicate the genome despite endogenous and exogenous obstacles. Replication origins are "licensed" in G1 for use in the next S-phase by loading the replicative helicase in an inactive form around DNA. During S phase, protein kinase and accessory factors cooperate to activate licensed origins and assemble bidirectional replisomes. Only a fraction of the licensed origins are activated during S phase. The remaining licensed origins are inactivated by passing forks during normal replication, although they can provide backup initiation events when fork progression from active origins is perturbed. Origins are activated at different times through S phase, which generates cell-type specific replication timing and replication direction profiles. Our long-term goal is to provide a quantitative description of replication origin firing and replication fork progression in eukaryotic genomes, including yeast and human cells. To achieve this goal, we are developing novel replication mapping techniques at both the cell population and the single molecule level and mathematical models of genome replication.
Recent results
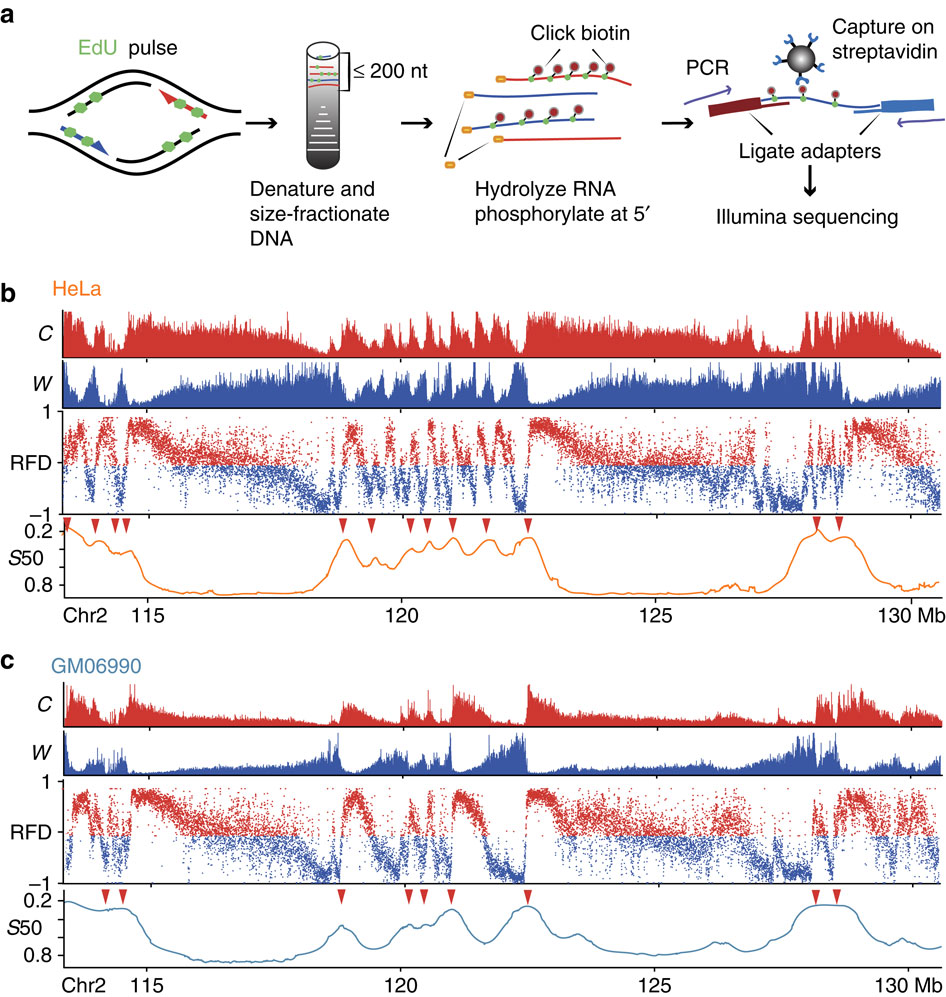
We mapped the direction of replication fork progression genome-wide in human cells by purification and strand-oriented sequencing of Okazaki fragments from cell populations (OK-seq) (1). Changes in replication fork direction revealed thousands of replication initiation and termination zones as well as unidirectionally replicating regions and randomly replicating regions. By profiling multiple cell lines, we identified genome compartments whose replication mode is constitutive or cell-type specific. We notably identified oncogene-repressed initiation zones in a model for chronic myeloid leukemia progression (2). We have confirmed master initiation zones mapped by OK-seq with an independent origin mapping technique called EdUseq-HU (3). This study also highlighted an interesting connection between preferential initiation sites and preferential sites of DNA breakage during S phase.
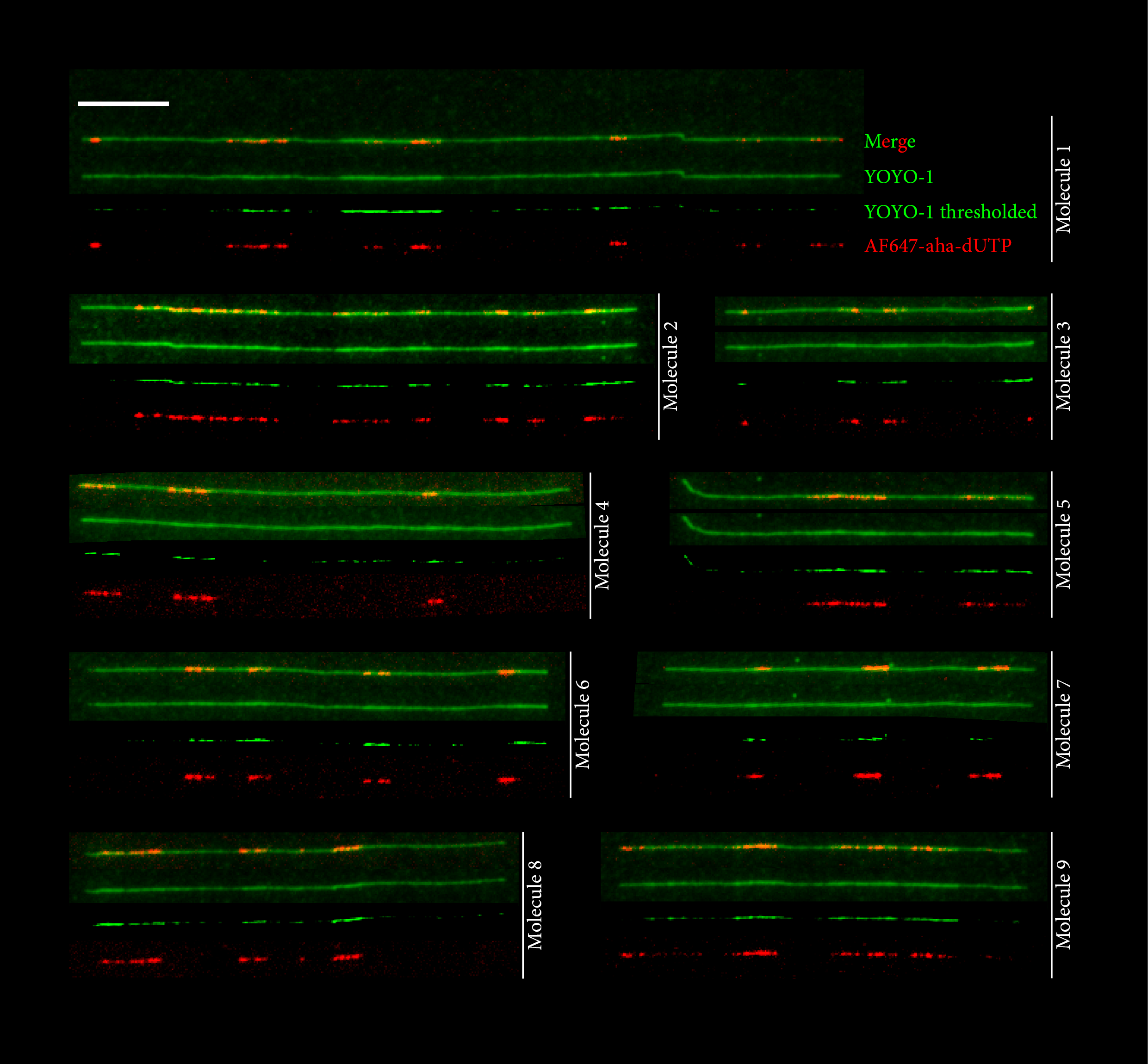
In addition to identifying efficient, "master" initiation zones, our results suggest that replication can also initiate inefficiently and dispersively between master origins. We obtained evidence for dispersed replication initiation between master initiation zones at a few exemplary loci using DNA combing, a classical technique to visualize the replication pattern of single DNA molecules (4). However, DNA combing is low-throughput and not scalable to the whole genome. We therefore combined fluorescent DNA precursors and nanochannel array technology to develop a high-throughput method for optical mapping of replicated DNA (HOMARD) in Xenopus egg extracts (5). An adaptation of this method to human cells by others revealed early-firing human IZs that corroborated OK-seq results (Wang et al. 2021). Nevertheless, resolution and informatics issues still restrain the power of optical methods.
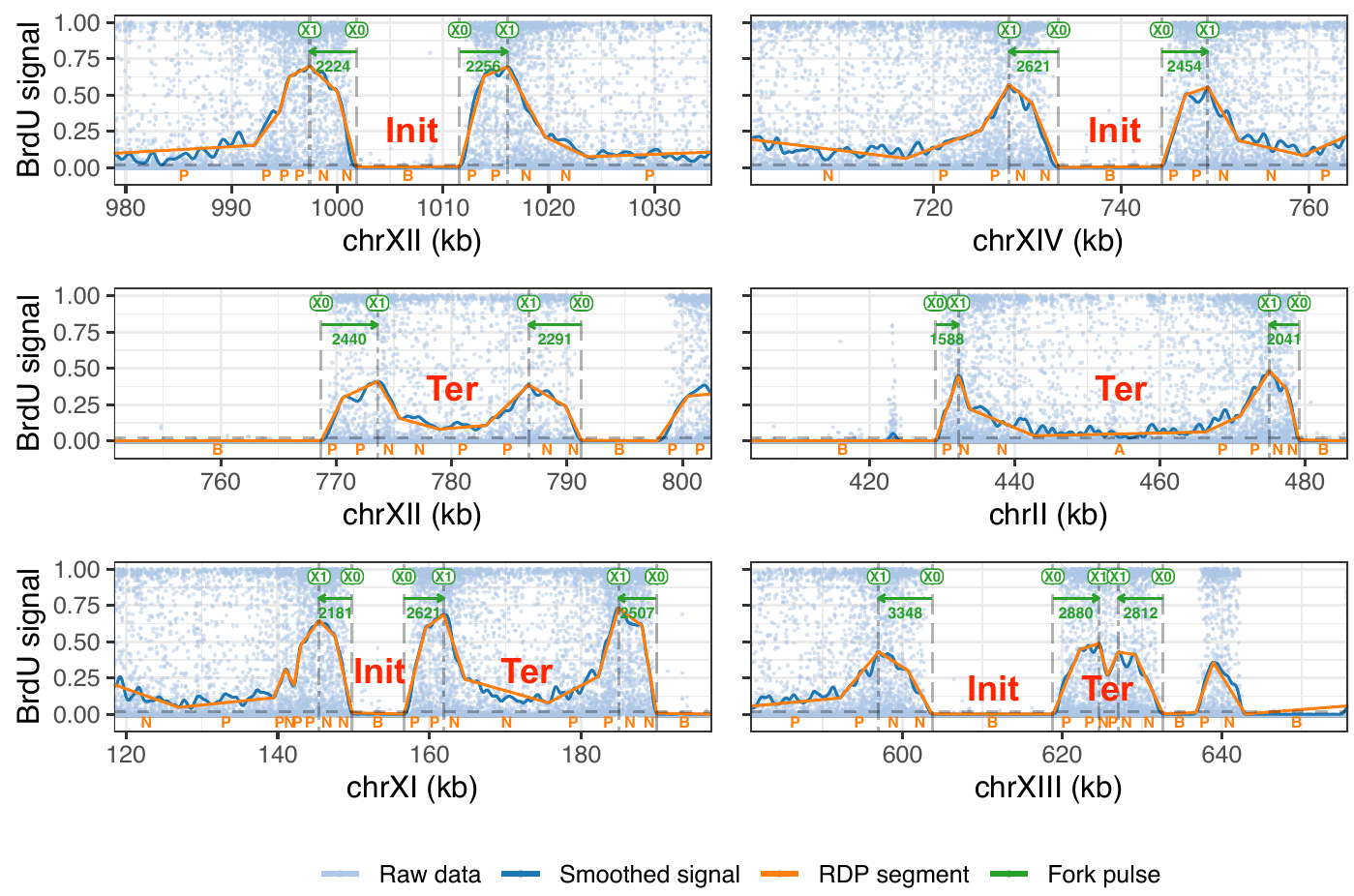
More recently, we developed FORK-seq, a nanopore-sequencing based method to map replication genome-wide at the single molecule level and at near-nucleotide resolution. The idea is to pulse-label replicating DNA of living cells with nucleoside analogs such as BrdU and to use nanopore sequencing of native DNA to reveal the patterns of analog incorporation and the genomic position of each sequenced molecule (6). Using artificial intelligence to precisely quantify BrdU incorporation, we detected and oriented 60,000 replication forks in the genome of the yeast S. cerevisiae. The results confirmed the known replication initiation and termination sites, but also revealed that 10% of initiation events occur in a dispersed manner, elsewhere than at previously known origins. We further developed NanoForkSpeed, which is additionally able to determine fork velocity from each labelled tract, and thus produced the first-ever genome-wide map of replication fork speed in a eukaryotic organism (7). We believe FORK-seq and NanoForkSpeed have huge potential for future DNA replication studies in many experimental systems and are currently adapting these techniques to human cells.
Cell-population and single-molecule data contain key information to mathematically model the DNA replication process. We previously demonstrated that the time-dependent rate of origin activation has a universal bell-shape in eukaryotes. We reproduced this shape by modeling genome replication as a probabilistic activation of helicases by limiting factors incorporated into replication forks (8). The universal shape emerges from a competition between origin activation and passive replication, predicting a novel mathematical relationship between initiation rate, fork speed and origin density that we could precisely verify in all tested eukaryotes. Using artificial intelligence, we extracted from genome-wide replication timing and directionality profiles an initiation probability landscape that, when inserted into this simulation framework, reproduced the experimental profiles with nearly complete accuracy. A comparison of the initiation probability landscape with experimental profiles of licensed origins showed that the density of licensed origins is not sufficient to predict genomic replication profiles (9,10). We propose that origin selection operates both at the origin licensing and the origin activation step, and are further investigating the genetic and epigenetic determinants of these two steps of replication origin regulation.
Current projects
DNA replication is a fundamental biological process that ensures the faithful transmission of genetic information through cell and organism generations. Perturbations of this process are a major source of DNA damage and genomic instability in cancer, aging and developmental diseases and are exploited by chemotherapies. FORK-seq (6), NanoForkSpeed (7) and a novel nanopore-sequencing based technique to map replication time, combined with genetics, artificial intelligence and mathematical modelling techniques, provide powerful novel tools to further explore the determinants of replication origin firing and replication fork progression and the impact of their perturbations in wild-type and mutant yeast and human cells. The results will broaden our understanding of this fundamental life mechanism and potentially guide novel therapeutic strategies.
Selected publications
1. Replication landscape of the human genome.
Petryk N, Kahli M, d’Aubenton-Carafa Y, Jaszczyszyn Y, Shen Y, Sylvain M, Thermes C, Chen CL, Hyrien O (2016). Nature Comm., 7, 10208.
2. Developmental and cancer-associated plasticity of DNA replication preferentially targets GC-poor, lowly expressed and late-replicating regions.
Wu X*, Kabalane H*, Kahli M, Petryk N, Laperrousaz B, Jaszczyszyn Y, Drillon G, Nicolini FE, Perot G, Robert A, Fund C, Chibon F, Xia R, Wiels J, Argoul F, Maguer-Satta V, Arneodo A, Audit B, and Hyrien O (2018),Nucleic Acids Res., Nov 2 ; 46(19):10157-10172. doi : 10.1093/nar/gky797. * joint first authors.
3. Human ORC/MCM density is low in active genes and correlates with replication time but does not delimit initiation zones
Nina Kirstein, Alexander Buschle, Xia Wu, Stefan Krebs, Helmut Blum, Elisabeth Kremmer, Ina M Vorberg, Wolfgang Hammerschmidt, Laurent Lacroix, Olivier Hyrien,Benjamin Audit, Aloys Schepers. Elife. 2021 Mar 8 ;10:e62161. doi : 10.7554/eLife.62161.
4. DNA molecular combing-based replication fork directionality profiling
Marion Blin, Laurent Lacroix, Nataliya Petryk, Yan Jaszczyszyn, Chun-Long Chen, Olivier Hyrien, Benoît Le Tallec. Nucleic acids research, 49(12), e69. https://doi.org/10.1093/nar/gkab219
5. High-throughput optical mapping of replicating DNA.
De Carli F*, Menezes N*, Berrabah W, Barbe V, Genovesio A, Hyrien O (2018)Small Methods, 2018, Sep 11 ; 2(9):1800146, doi : 10.1002/smtd.201800146. *joint first authors.
6. FORK-seq : replication landscape of the Saccharomyces cerevisiae genome by nanopore sequencing
Hennion M, Arbona JM, Lacroix L, Cruaud C, Theulot B, Le Tallec B, Proux F, Wu X, Novikova E, Engelen S, Lemainque A, Audit B, Hyrien O (2020) Genome Biol., 2020 May 26 ;21(1):125. doi : 10.1186/s13059-020-02013-3.
7. Genome-wide mapping of individual replication fork velocities using nanopore sequencing
Bertrand Theulot*, Laurent Lacroix*, Jean-Michel Arbona, Gael A. Millot, Etienne Jean, Corinne Cruaud, Jade Pellet, Florence Proux, Magali Hennion, Stefan Engelen, Arnaud Lemainque, Benjamin Audit, Olivier Hyrien & Benoît Le Tallec. Nature communications, 13(1), 3295. https://doi.org/10.1038/s41467-022-31012-0 *joint first authors
8. The eukaryotic bell-shaped temporal rate of DNA replication origin firing emanates from a balance between origin activation and passivation.
Arbona JM, Goldar A, Hyrien O, Arneodo A, Audit B. Elife. 2018 ;7:e35192. Published 2018 Jun 1. doi:10.7554/eLife.35192
9. Human ORC/MCM density is low in active genes and correlates with replication time but does not delimit initiation zones
Nina Kirstein, Alexander Buschle, Xia Wu, Stefan Krebs, Helmut Blum, Elisabeth Kremmer, Ina M Vorberg, Wolfgang Hammerschmidt, Laurent Lacroix, Olivier Hyrien,Benjamin Audit, Aloys Schepers. Elife. 2021 Mar 8 ;10:e62161. doi : 10.7554/eLife.62161.
10. Neural network and kinetic modelling of human genome replication reveal replication origin locations and strengths
Jean-Michel Arbona, Hadi Kabalane, Arach Goldar, Olivier Hyrien, Benjamin Audit. bioRxiv 2021.12.15.472795 ; doi : https://doi.org/10.1101/2021.12.15.472795